
Paroxysmal Nocturnal Hemoglobinuria - A dark mystery
A hundred and eleven years.
That is the time that it took to understand the curious case of Paroxysmal Nocturnal Hemoglobinuria.
True to the testament of how evidence based medicine should work, the mystery of PNH was described sequentially from the symptom, discernable to the naked eye and zoomed into the gene that is proposed to cause this particular problem, by sheer perseverance, curiosity and the constant search of answers.
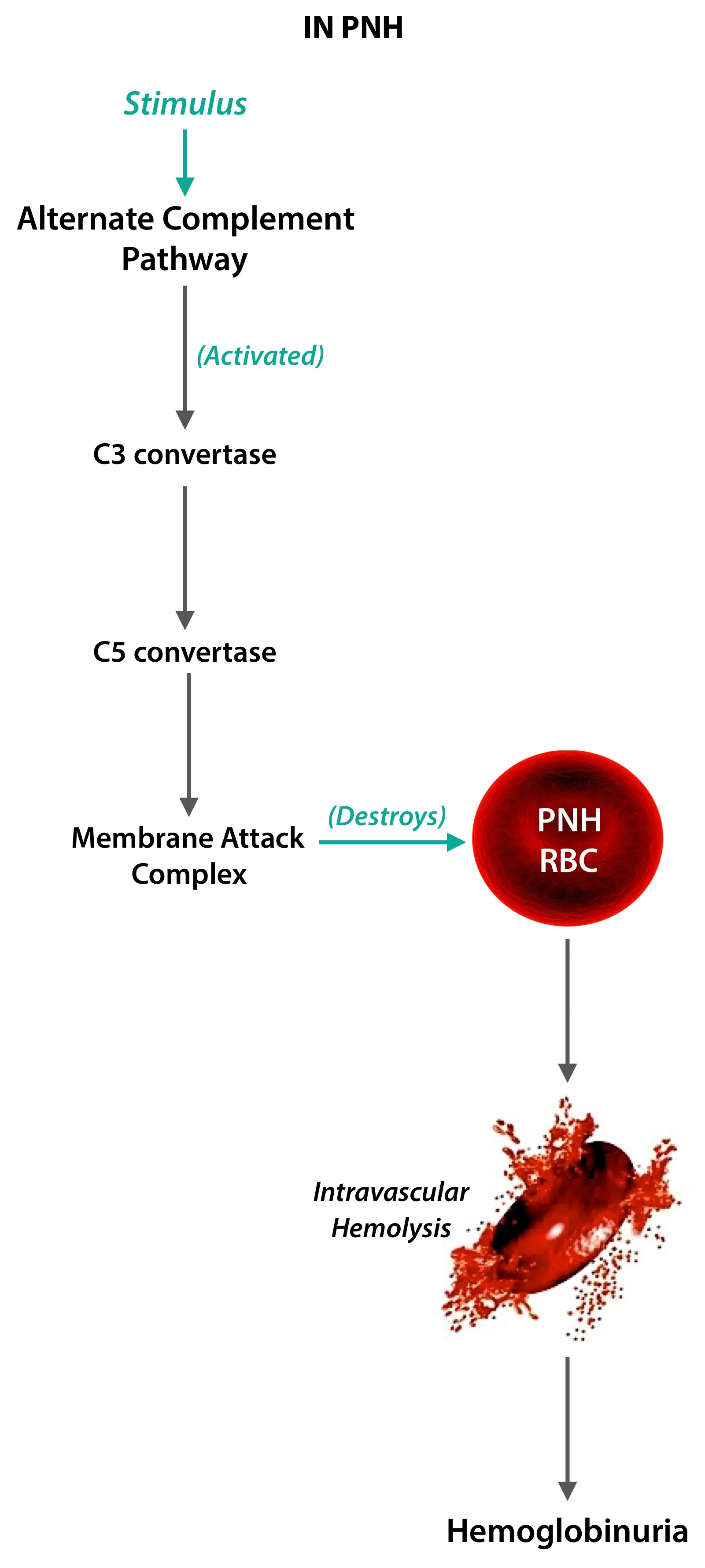
The typical patient having PNH will come with a peculiar complaint of passing dark urine as soon as he or she wakes up in the morning, a particularly distressing complaint to have. This is because of destruction of RBCs in the blood vessels, and the consequent hemoglobinuria that occurs.
At this point, we are thinking..
“How are the RBCs just getting destroyed for no apparent reason? Why is this hemolysis occurring only in the night?”
When we sleep, by virtue of some level of hypoventilation, there is a little bit of carbon dioxide retention in our body. Carbon dioxide is an acidic molecule; hence the pH of our blood becomes more acidic. Normally, this shouldn’t matter, but in someone with PNH, this lowered pH causes RBC destruction that is pronounced at night, and the patient wakes up and passes dark coloured urine. This was proved by studies where the RBCs of patients with PNH lysed in a simulated acidic environment, whereas the normal RBCs withstood this acid challenge, thus paving way for the diagnostic tests used in the past; the Ham’s test and the Sucrose lysis test.
So, it was established that some unknown entity was making these RBCs prone to destruction by an acidic medium.
The “why?” was answered on subsequent discovery of the alternate complement system.
The alternate complement pathway is activated by many mechanisms, most commonly by bacterial cell wall components, and some plasma proteins; acidic pH enhances the complement system’s efficiency. Normally, our body’s own cells have many mechanisms by which they evade killing by our immune system, by a process called immunological tolerance. We had learnt about this concept in the “Sympathetic Ophthalmitis” article.
After decades research, it was understood that the PNH RBCs were abnormally susceptible to complement mediated lysis. This doesn’t occur in normal individuals. So presumably, there is some part of the puzzle that makes these RBCs susceptible.
Central to the pathophysiology of PNH, happens to be an acquired mutation in a gene that encode for molecules that prevent complement mediated lysis. These are called GPI anchor molecules which are coded by the PiG-A gene on the X-chromosome. Men have only one X-chromosome and women have only one of the two X-chromosomes functioning at any given time (by a mechanism called lyonisation) and if there is a mutation, it’s purely by chance (no two people have the same PiG-A gene mutation). It will manifest in the affected person, but luckily the disease will not be transmitted the future generations.
Okay, so why is this tiny gene so important to us? Turns out, this gene has the information to produce anchor proteins that prevent the complement from destroying the cells; most important of which are the Decay Accelerating Factor (DAF) and the Membrane Inhibitor of Reactive Lysis (MIRL).
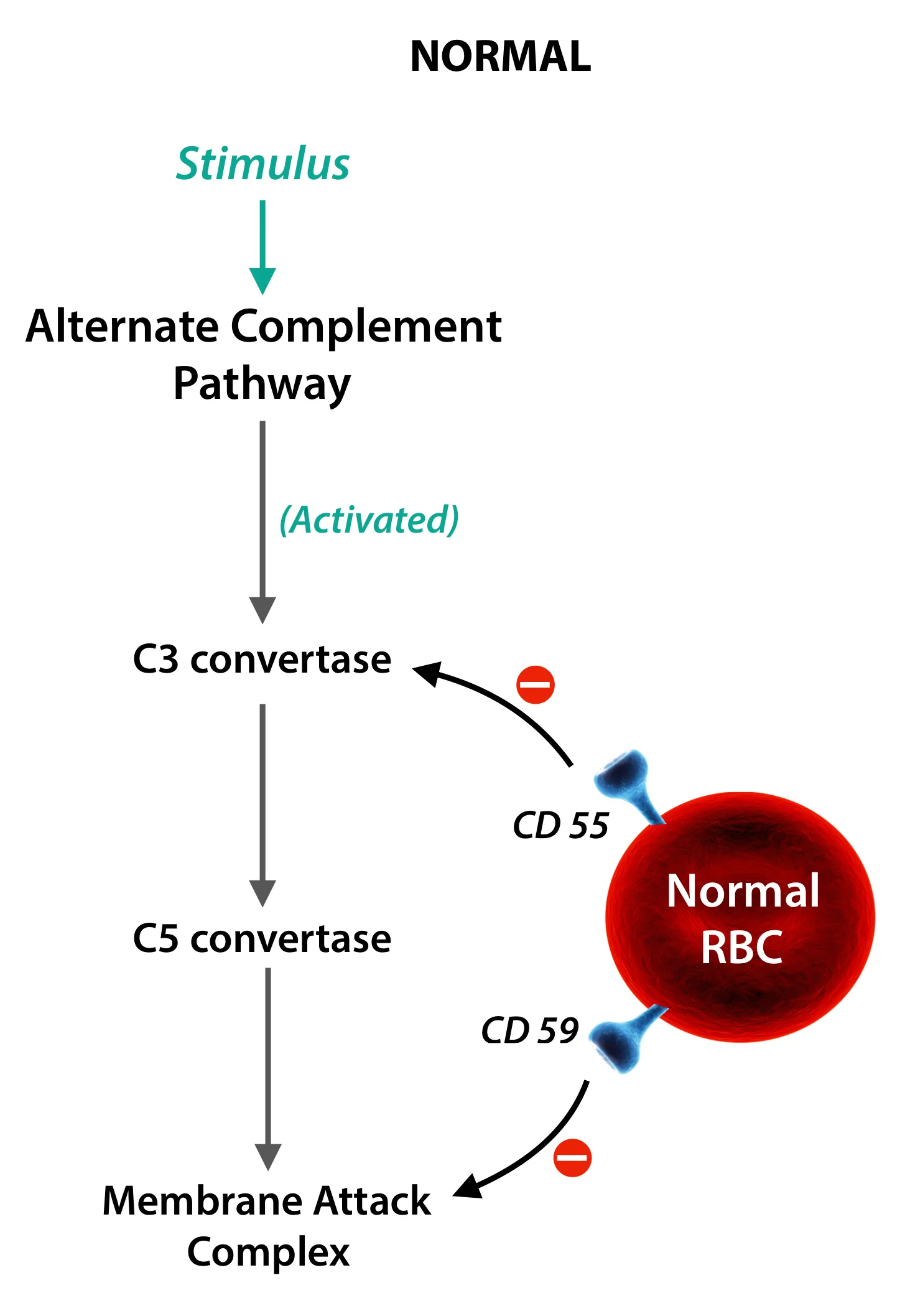
So, No anchor molecule = No attachment of protective factors = No protection = RBC destruction.
These molecules, the DAF and MIRL have specific cell surface markers, CD 55 and CD 59 respectively; and on performing a test called the Fluorescent In-Situ Hybridisation, popularly called FISH, they are characteristically found to be absent, which clinches the diagnosis of PNH.
There is curious relationship to PNH and aplastic anemia, a bone marrow failure syndrome that causes pancytopenia (low cell counts of RBCs, WBCs and Platelets). One theory is that this PiG-A gene mutation occurs in the hematopoietic stem cells, thus making all cell lines susceptible to complement mediated lysis, hence causing pancytopenia.
The other theory is a little more interesting; it’s been said that this PiG-A mutation is seen in low levels in normal population. The normally functioning marrow ensures that this is an insignificant occurrence. But in the setting of bone marrow failure, the cells with this PiG-A mutation undergo clonal expansion, leading to PNH. Which among the two is the cause and the effect is up for debate, and intense research into finding the reason for the clonal selection may give answers to many other conditions of the hemotological system.
Despite all this, the reality is that Paroxysmal Nocturnal Hemoglobinuria is quite rare. Though better modalities of diagnosis and treatment of this condition have come up, the story of the ongoing investigation into the basis of this disease reminds us that knowingly or unknowningly, every single one of us in this world are working towards a common goal- better understanding of this curious thing called the human body.
But wait. Why is the urine dark only in the early morning as explained earlier? Though the hemolysis occurs throughout the day, the hemolysis is more pronounced when we sleep by virtue of the pH changes and it has been named Paroxysmal Nocturnal Hemoglobinuria for this precise reason; paroxysms of hemoglobinuria occurring overnight.
Author: Anirudh Murali (Facebook)
Sources and citations
"Historical Aspects of Paroxysmal Nocturnal Haemoglobinuria: 'Defining the Disease'." British Journal of Hematology 1.117 (2002): 3-22. Web.
Luzzatto, Lucio. "Chapter-129: Hemolytic Anemias and Anemias Due to Acute Blodd Loss."Harrison's Principles of Internal Medicine. 19th ed. N.p.: McGraw - Hill Education, 2015. 660-62. Print.
Kumar, Vinay, et al. “Chapter-2: Inflammation and Repair.”Robbins Basic Pathology, 9th ed., Elsevier-Saunders, 2013, pp. 50–51.